As mucopolissacaridoses (MPS) formam um grupo de distúrbios metabólicos hereditários caracterizados pelo armazenamento intralisossomal de glicosaminoglicanos (GAGs) secundário a uma deficiência na atividade de uma enzima lisossômica específica. Esse armazenamento anormal compromete tanto a arquitetura quanto a função de células e órgãos e resulta em um amplo espectro de manifestações clínicas progressivas e multissistêmicas.
As manifestações oftalmológicas da MPS incluem pseudoexoftalmia com fácies, grosseira característica, órbitas rasas, ptose, opacidade da córnea, glaucoma, papiledema por hidrocefalia ou compressão ou infiltração do nervo óptico, atrofia óptica e retinopatia.
Estudos histopatológicos documentaram acúmulo progressivo de GAG em várias estruturas oculares, incluindo conjuntiva, córnea, íris, cristalino e esclera.
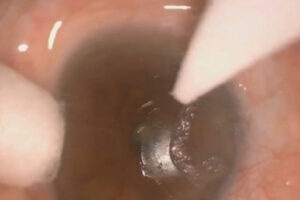
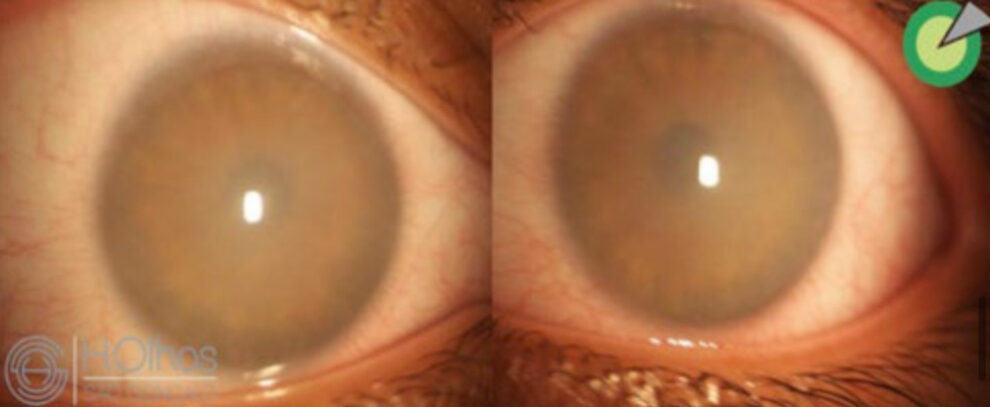
Neste relato de caso, apresentamos uma paciente jovem, portadora de MPS tipo VI, com hipertensão ocular e baixa acuidade visual secundária a opacidade corneana bilateral, que foi submetida a transplante lamelar anterior profundo (DALK) para reabilitação visual.
Relato de caso
Paciente M.C.S.M, feminina, 15 anos, portadora de MPS VI, tratada com reposição lisossomal, apresenta queixa de piora da acuidade visual e baixo desempenho escolar. Ao exame oftalmológico, 29 de maio de 2019, apresenta melhor acuidade visual corrigida (MAVC) de 20/200 em ambos os olhos, mobilidade ocular extrínseca preservada, pressão intraocular (PIO) 38 mmHg em olho direito (OD) e 37 mmHg em olho esquerdo (OE). O exame biomicroscópico revelou opacidade corneana estromal difusa bilateral, impossibilitando a realização de fundoscopia e a avaliação detalhada do segmento anterior.
Clique aqui para ler o artigo na íntegra
Autora

INAÊ SAMPAIO
Residente do terceiro ano de oftalmologia do H. Olhos São Gonçalo.